JA
JA
 中文
中文




Miriam Coelho Molck Milena Simioni Társis Paiva Vieira
Fabíola Paoli Monteiro Vera L. Gil-da-Silva-Lopes
カンピナス大学医学部遺伝学科(UNICAMP)、ブラジル、カンピナス
確定事実
報告された10q22.3q23.2の微小欠失は、最近報告されたまれな10q22q23欠失症候群と関連しており、特徴的ではない表現型を示す。
現在までに、10q22.3q23.2に隣接する低コピー反復3/4における14例の微小欠失が文献で報告されている。
新しい洞察
10q22.3q23.2で7.8Mbの微小欠失を呈した男児の新規症例を報告し、文献で報告された類似症例と彼の臨床徴候を比較した。
さらに、著者らの患者は、表現型に関連する可能性のある189 kbの16q12.1の微小欠失を有している。
キーワード
欠失症候群10q22q23 ・ 遺伝子型-表現型相関 ・ 分子細胞遺伝学
要約
10q22.3q23.2領域の欠失はまれであり、2つの低コピー反復(LCR3および4)によって媒介される。 これらの欠失はすでに10q22q23欠失症候群として認識されている。 この病態に関連する表現型はかなり特徴的ではなく、最も一般的な特徴は頭蓋顔面の異形および発達遅延である。 頭蓋顔面奇形の特徴、発達遅滞、ファロー四徴症、手足の異常、および再発性気道感染症を有する男児について報告する。 染色体マイクロアレイ分析は、10q22.3q23.2で7.8Mbの微小欠失、LCRs 3/4に隣接し、さらに189 kbの16q12.1の微小欠失を示した。 本稿では、類似の欠失を有する報告症例の臨床徴候をレビューし、それらを我々の患者と比較し、遺伝子型‐表現型相関のより良い理解に貢献した。
著作権(2017 S. Karger AG, Basel)
分子細胞遺伝学的解析の向上。MLPAや染色体マイクロアレイを用いた分子細胞遺伝学的解析の向上により 染色体マイクロアレイを用いた分子細胞遺伝学的解析の向上により、微小欠失/微小重複症候群のリストは増加の一途をたどっています。 microduplication syndromesのリストは常に増加している[Slavotinek, 2008; Nevadoら 2014]。これらの中には、認識可能な表現型を持つものもあれば、表現型が一貫しておらず、神経精神疾患のリスクを高めるものもあります[Koolenら、2008;van Bonら、2011]。
10q22.3q23.2の欠失は、認知障害および行動異常に関連するまれな新規症候群として特徴づけられている[Balciunieneら、2007]。 この領域は、非対立遺伝子相同組換え(NAHR)[Lupski と Stankiewicz, 2005]によって媒介される様々なゲノム変化を引き起こすことができる、設計されたLCR3およびLCR4である複雑な低コピー反復セット(LCR)に隣接している。
この領域にLCRが存在することは、この遺伝子座が染色体再編成に対する感受性が高いことを示唆している[Allimanら、2010]。 それにもかかわらず、LCR3/4内の切断点を有する14個の新規欠失および7個の重複のみが記載されている[Gossら 1998; Hanら 2004; Dufkeら 2006; Balciunieneら 2007; Erdoganら 2008; Allimanら 2010; Reddyら 2011; Singhら 2011; van Bonら 2011; Petrovaら 2014]。
本研究では、10q22.3q23.2で7.8 Mbの微小欠失を有し、LCR3/4に隣接し、さらに16q12.1で189kbの微小欠失を有する患者について報告する。
患者と方法
症例報告
3歳の少年が、複数の異常のため、FCM-UNICAMPの遺伝学サービスで遺伝医学コンサルテーションのため紹介された。 患者は経験豊富な臨床遺伝学者により、臨床歴および家族歴情報と異形の特徴からなる特定のチェックリストを用いて評価された。
発端者は、自然流産の既往があり、年長の健康な子供をもつ血縁関係のない両親から生まれた2番目の子供です。 彼の家族歴は特記すべきものではなかった。 出生時、両側内反足が認められた。 新生児期にFallot四徴症と診断され、9か月齢で矯正心臓手術を行った。 その後の発達は全般的に遅れ、小児期に1回の熱性痙攣が報告された。 また、上気道感染の再発が認められた。
3歳時の形態学的検査では、前頭隆起、両側の過度に折り畳まれた螺旋と顕著な右反耳輪を伴う低位前捻耳、右耳介前窩、まばらな眉毛、下斜めの眼瞼裂、浅眼窩、鼻の球根状の先端、高口蓋、両側内反足、両側の不完全な手掌裂が明らかとなった。
分子細胞遺伝学的分析
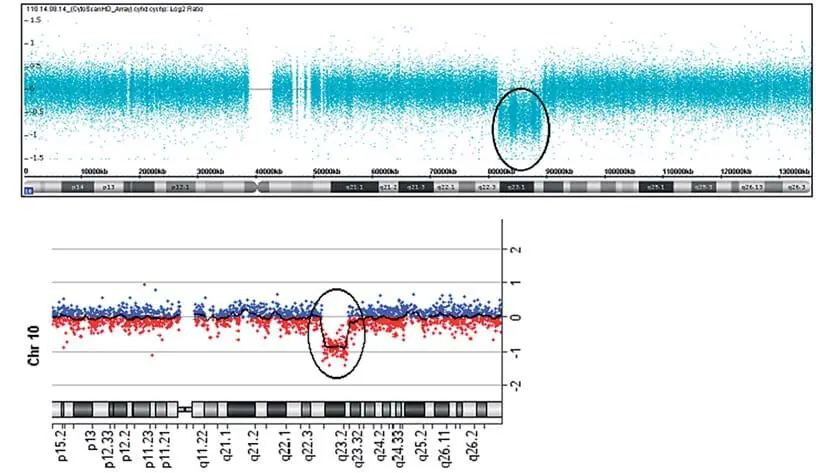
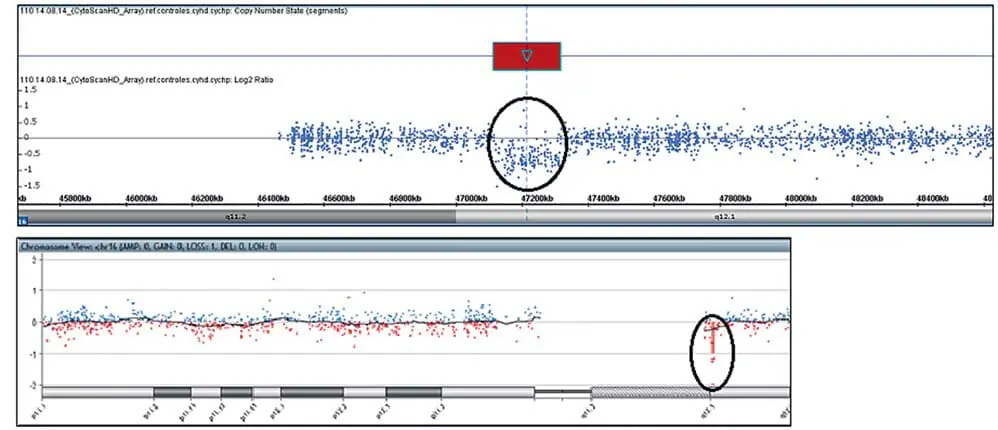
CytoScan HDチップ(Affymetrix, Santa Clara, CA, USA)を用いて、製造者の指示に従って染色体マイクロアレイ分析を行い、染色体分析スイート(ChAS)ソフトウェア(Affymetrix)を用いて分析した。 病原性CNVの確認は、Agilent Human Genome G3 SurePrint 8 × 60 K Microarray(Agilent Technologies, Santa Clara, CA, USA)を用いて行い、Agilent Cytogenomicsソフトウェア(Agilent Technologies)を用いて分析した。 患者の染色体マイクロアレイ分析は、10q22.3q23.2で7.8 Mbの微小欠失を明らかにした(図1A,B)。 さらに、著者らの患者は16q12.1(図2A、B)で189kbの微小欠失を有していた:arr[GRCh37]10q22.3q23.2(8146577_89253430)×1,16q12.1(47127281_47316339)×1。 これらの欠失の親起源の調査は、試料が入手できなかったため不可能であった。 患者は追跡不能となり、核型は構造的変化を除外するために実施できなかった。
考察
発達遅延、先天性心疾患(CHD)、頭蓋顔面異形症、手足の異常、および再発性感染症を有する男児の症例を報告する。 実際、患者は、稀な10q22q23欠失症候群(DS)の領域に重複する7.8Mbの10q22.3q23.2間質性微小欠失を呈する。 報告された患者およびLCR3/4 10q22.3q23.2欠失を有する発端者の一般的な臨床的特徴には、全15例に認められる頭蓋顔面異形の特徴、発達遅延(13/15、87%)、口蓋の変化(6/15、40%)、先天性心疾患(6/15、40%)、および手足の異常(10/15、67%)が含まれた。 我々の患者には存在しない10q22q23 DSのその他のまれな臨床的特徴は、筋緊張低下(3/15、20%)、自閉症(2/15、13%)、関節伸展性亢進(2/15、13%)である(表1)。 頭蓋顔面の異形は、認識可能な顔面の表現型をもたらさず、最も一般的には低眼瞼症または高眼瞼症(9/15、60%)、低耳および小耳(6/15、40%)、上向きまたは下向きの眼瞼裂(6/15、40%)、上向きのひだ(4/15、27%)、および平坦な鼻橋(3/15、20%)を含んでいた。 著者らの患者は、低位耳と下向き眼瞼裂を呈した。 最も一般的な口蓋の変化は、15例中5例(33%)に認められる高口蓋であり、これには本稿で報告したものも含まれる。 最も頻繁に観察される先天性心疾患は、患者15人中3人(20%)で同定された心室中隔欠損であるが、Fallot四徴症は我々の患者では認められなかった。 手足の異常には、一般的に内反足(4/15、27%)が含まれており、我々の患者にも認められた。くも膜指症(2/15、13%)も含まれていた。 本患者で観察された別の臨床的特徴は、再発性上気道感染であり、他の10q22q23 DS症例では報告されなかった。 著者らの患者で検出された10q22.3q23.2の7.8Mb欠失領域内に16のOMIM遺伝子がある。 これらの遺伝子のいくつか、例えば、BMPR1A(601299)、LDB3(605906)、GRID1(610659)、およびNRG3(605533)は、表現型に関連する推定候補遺伝子として、特に神経心理学的発達および心臓欠損に関して仮定されている[Balciunieneら 2007; van Bonら 2011; Breckpotら 2012]。 斎藤ら [2012]神経堤由来細胞における変異マウスノックアウトBMPR1A蛋白質の心臓形態を評価し、それらのいくつかは心臓欠損以外に顔面裂や口蓋裂などの顔面融合欠損、または顔面異形特徴を示すことを示した。 このように、著者らの患者で観察された頭蓋顔面異形および先天性心疾患は、BMPR1A遺伝子の欠失によって部分的に引き起こされた可能性がある。
BMPR1A遺伝子のヘテロ接合性点突然変異または部分欠失は、胃腸管に過誤腫性ポリープが発生することを特徴とする若年性ポリポーシス症候群(OMIM 174900)とも関連している[Larsen と Howe, 2017]。 10q22.3q23.2欠失を有する患者のうち、乳児若年性ポリポーシス症候群と呼ばれる若年性ポリポーシス症候群の1つのサブタイプは、BMPR1AおよびPTEN遺伝子の両方が欠失している患者にのみ観察されている[Menkoら、2008]。 PTENは、認識された腫瘍抑制因子の役割[Liら 1997]を有し、カウデン症候群およびBannayan-Riley-Ruvalcaba症候群と関連しており、ポリポーシス発症にはBMPR1AおよびPTENの両方の連続的欠失が必要であることを示唆している[Zhouら 2003; Dahdalehら 2012]。 PTENは、LCR 3/4側面10q22.3q23.2欠失では存在せず、同様の欠失を有する患者では、乳児期の若年性ポリポーシスまたはポリポーシス表現型を発現したと報告された患者はいなかった[Petrovaら 2014]。
LDB3は、細胞骨格アセンブリに関与するLIM結合ドメイン3タンパク質をコードし、この遺伝子の突然変異は、ヒトにおける拡張型心筋症と関連している[Vattaら 2002; Hershbergerら 2008]。 心臓の機能または構造におけるこのタンパク質の影響は、研究中である[Alimanら、2010]。
GRID1遺伝子はグルタミン酸受容体チャネルのサブユニットをコードしており、中枢神経系における興奮性シナプス伝達を媒介する役割を担っている[Yamazakiら、1992]。 Vasanら [2009]は、心臓構造と機能に関連する共通の遺伝的変異体のゲノムワイド関連メタアナリシスを通して、左室壁厚の候補遺伝子としてGRID1を提案した。 さらに、この遺伝子は、いくつかの研究[Fallinら 2005; Coyle, 2006; van Bonら 2011]でも統合失調症と関連している。 GRID1の近位に位置し、統合失調症のリスクおよび症状と関連していると報告されている別の遺伝子は、NRG3[Chenら 2009]である。NRG3は、神経系、心臓、乳房、および他の器官系における細胞間相互作用を媒介するシグナル伝達タンパク質において役割を果たしている[Falls, 2003]。
さらに、著者らの患者は16q12.1(189 kb)に微小欠失を有している。 この領域は2つのOMIM遺伝子: NETO2(607974)およびITFG1(611803)を含む。 NETO2はカイニン酸受容体サブユニットであり、脳内のグルタミン酸シグナル伝達機構に有意な影響を及ぼすと考えられている[Zhangら 2009]。 ITFG1は、提唱されている細胞接着機能を有する膜貫通タンパク質である[Katoら、2014]。 この欠失は、DGVデータベース(http://projects.tcag.ca/variation)の個人では検出されなかった。 DECIPHERDatabase(https://decipher.sanger.ac.uk/),、本症例で認められた欠失を包含するはるかに大きな欠失)では、2例(2856例および289229例)に認められた唯一の所見として記載されており、本症例と共通した発達遅延が認められた。 また、DECIPHER由来の症例289229は心房中隔欠損である先天性心疾患を呈した。 この欠失が表現型に及ぼす重要性を明らかにするためには、類似の欠失をもつ他の個体についてのより詳細な情報、特にこれらの遺伝子の特異的な機能についてのより詳細な情報が必要である。 さらに、10q22q23 DS患者15例中3例に染色体異常が認められ(表1)、表現型に影響を及ぼした可能性がある。 最近、大きなゲノムスケールの構造的変異体、すなわち、DNAの巨大塩基を含む変化は、ワンオフ事象(クロモアナシンセシスおよびクロモトリプシス様事象)において生成される他の構造的変異体、またはヘテロ接合性の欠如を伴う複雑なゲノム再編成など、さらなる複雑性と関連し得ることが示唆されている[Carvalo と Lupski, 2016]。 異なる疾患表現型は異なる器官系に影響を与えるが、重複する疾患表現型は同じ経路内で相互作用するタンパク質をコードする2つの遺伝子によって引き起こされる可能性が高い。 複数の遺伝子における病原性変異体間のこの相互作用は、1人の患者内で表現型に関して観察される複雑なスペクトルをもたらし、関連するゲノム障害の可変性に寄与する[Poseyら 2017]。
我々の患者に認められた臨床徴候のほとんどは、まれな10q22q23 DSで報告されている。 しかしながら、我々の患者およびこの欠失を有する他の患者における知見は、コア表現型の特徴付けを可能にしない。 さらに、表現型に対する16q12.1欠失の影響を排除することはできない。
若年患者におけるまれで新しいゲノム不均衡の同定は、行動異常および発達遅延に対する早期介入を可能にすることによって臨床管理を変えることができ、また特異的な追跡を確実にする。 さらに症例を追加すれば、遺伝子型-表現型相関およびこの疾患の病因に対する複数の稀な変異体の効果が改善されるであろう。
表1 LCR3/4に隣接する10q22.3q23.3欠失を有する患者の臨床的特徴
| 参考文献、症例 | 削除サイズ | 相続 | 性別 | 評価時の年齢 | 発達遅延 | 先天性心疾患 | Palateの変更 | 顎顔面の異形性 | 手足の変化 | その他の臨床的特徴 | その他の奇形 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| バルシユニエン他 [2007] | |||||||||||
| UM10qDel-01 | 7.5 Mb | de novo | 男性 | 3歳6か月 | + | – | – | 軽度の特徴 | – | 自閉症 | – |
| JHU10qDel-01 | 7.5 Mb | de novo | 男性 | 1歳6か月 | + | – | – | 白色前ロック | – | 小脳後部、小嚢胞、小脳 | – |
| アリマンら [2010] | |||||||||||
| 症例1 | 7.25 Mb | de novo | 男性 | 2歳7か月 | + | 動脈管開存 | 高弓状口蓋 | 軽度の高色素症、立ち上がった眼瞼裂、外側耳介の過剰な折り畳み、耳葉のしわ、細い上唇、小顎症 | クモ指症 | 自閉症、関節の伸展性亢進 | – |
| 症例2 | 7.25 Mb | de novo | 女性 | 17歳1か月 | + | – | 高弓状口蓋 | 軽度の顔面非対称、下向きの眼瞼裂、境界線上のセット耳、頬の平坦化、突出 | 軽度のクモ指症 | 軽度の筋緊張低下、関節の伸展性亢進、近視の病歴、注意欠陥/過活動障害 | – |
| 症例3 | 7.25 Mb | de novo | 男性 | 8日間 | NA | – | – | 軽度の斜頭症、高眼瞼症、軽度の上眼筋ひだ、低位、後方回転耳、狭い外耳道を伴う正常頭症 | 両側内反足 | 軽度の筋緊張低下、両側性難聴 | – |
| 症例4 | 7.25 Mb | de novo | 男性 | 1歳8か月 | + | – | 高弓状口蓋 | 巨頭症、前頭部ボッシング、狭い真実、顎後症、小口、下斜めの眼瞼裂、高色素症、小後耳、両側切痕中間螺旋 | – | 軽度の筋緊張低下、発育不全 | – |
| van Bonら [2011] | |||||||||||
| 患者1 | 7.2 Mb | de novo | 女性 | 22歳以上 | + | – | – | 末梢機能低下、起立性眼瞼裂、眼瞼下垂、低位小耳 | 広母指と母趾 | 乳房形成不全 | – |
| 患者2 | 7.2 Mb | de novo | 女性 | 2歳6か月 | + | 心房室中隔欠損 | – | 高眼瞼症、前捻鼻孔、平坦な鼻橋、大きな口、テレカンサス、耳介低位 | – | – | – |
| 患者3 | 7.7 Mb | de novo | 男性 | 3歳7か月 | + | – | 高弓状口蓋 | ドリコセファリー、高眼瞼症、眼瞼上ひだ、平坦な鼻橋、顔面中央の陥没、耳介低位 | 後置母指 | 悪性歯、キアリI奇形、てんかん | de novo 2q36.3複製(722 kb) |
| 患者4 | 7.7 Mb | de novo | 男性 | 12歳 | + | TR、肺動脈弁逆流 | – | 長顔、ハイパーロリズム、アーモンド形の眼 | 橈尺骨癒合、側弯、後弯、漏斗胸 | カフェオレ斑点 | 47,XYY |
| 患者5 | 7.5 Mb | 母親由来 | 男性 | 5歳 | + | – | – | 高眼瞼症、鼻の広い底部、平坦な鼻橋、耳介低位 | 両側内反足 | 陰嚢をショールする | – |
| Reddyら [2011] | |||||||||||
| 症例2 | 7.7 Mb | de novo | 女性 | 4歳 | + | – | – | 幅広の鼻、反転した下唇、緑色の境界 | – | – | – |
| Singhら [2011] | |||||||||||
| 症例報告 | 7.46 Mb | de novo | 女性 | 5歳6か月 | + | 心房心室中隔欠損 | – | 軽度の天蓋裂、右方向の外斜視 | 角質増殖症、第5趾の爪 | 持続性、頻脈性の呼吸、再発性中耳炎 中耳炎 | – |
| Petrovaら [2014] | |||||||||||
| 臨床報告 | 7.3 Mb | de novo | 男性 | 2歳 | – | 心房心室中隔欠損、持続性卵円孔 | 正中口蓋裂 | 高眼瞼症、高額、広範な鼻橋、立ち上がった眼瞼裂、上顎骨ヒダ、顎後部、舌下垂 | 右側内反足 | – | – |
| 現在の症例 | 7.8 Mb | 不明 | 男性 | 3歳 | + | ファロー四徴症 | 高弓状口蓋 | 前頭突起、低位でわずかに前捻した耳介、両側の過度に折りたたまれた耳輪、顕著な右反耳輪、右耳介前窩、軽度のまゆ、下斜めの眼瞼裂、浅眼窩、鼻球先端 | 両側内反足、両側不完全掌側しわ | 再発性上気道感染 | 16q12.1欠失(189 kb) |
謝辞
我々は、ブラジル、サンパウロのカンピナスに所在するカンピナス大学医学部分子遺伝学研究室における患者とその家族の協力と、マルチユーザ装置の研究室に対し、装置、GeneChip Fluidics Station、GeneChip Scannerを用いた支援に感謝する。
本研究は、FAPESP – Fundaco de Amparo to Pesquissa do Estado Sano Paulo (2008/10596-0, 2009/08756-1, 2011/23794-7, 2012/51799-6)およびCNPq – Conselho Desolvimento Cientifico Technologico (149600/2010-0, 471422/2011-8)の支援を受けた。 V.L.G.-d.-S.-L.はCNPq(30455/2012-1)によって支持されている。
倫理宣言
本試験はカンピナス大学倫理委員会(No)の承認を受けた。 487/2009および433/2010。 患者の両親から文書によるインフォームド・コンセントを得た。 開示声明著者は、この記事の研究、著者権、および/または出版に関して、潜在的な利益相反はないと宣言している。
参考文献
Alliman S, Coppinger J, Marcadier J, Thiese H, Brock P, et al: 10q22.3q23.2における再発性ゲノム障害患者の臨床的および分子的特徴付け。 Clin Genet 78: 162-168 (2010)
バルシユニエンJ、フェンN、イヤドゥライK、ヒルシュB、シャルナスLら:再発性10q22-q23欠失:認知および行動異常に関連する10q上のゲノム障害。 Am J Hum Genet 80: 938–947 (2007).
Breckpot J, Tranchevent LC, Thienpont B, Bauters M, Troost E, et al: BMPR1Aは、再発性10q22q23欠失症候群に関連する先天性心疾患の候補遺伝子である。 Eur J Med Genet 55: 12–16 (2012).
Carvalho CM, Lupski JR: ゲノム障害における構造的変異体形成の根底にある機序。 Nat Rev Genet 17: 224–238 (2016). Chen PL, Avramopoulos D, Lasseter VK, Mc-Grath JA, Fallin MD, et al: 染色体10q22-q23上のファインマッピングは、統合失調症におけるニューレグリン3を意味する。 Am J Hum Genet 84: 21–34 (2009).
コイルJT:グルタミン酸と統合失調症:ドパミン仮説を超えている。 Cell Mol Neurobiol 26: 365–384 (2006).
Dahdaleh FS, Carr JC, Calva D, Howe JR: 染色体10q22-23の微小欠失を伴う若年性ポリポーシスおよび他の腸ポリポーシス症候群。 Clin Genet 81: 110-116 (2012)
Dufke A, Singer S, Borell-Kost S, Stotter M, Pflum DA, et al: De novo 構造的染色体不均衡: 部分的トリソミーの分子細胞遺伝学的特徴づけ。 細胞遺伝学ゲノム研究114:342-350(2006)。
Erdogan F, Belloso JM, Gabau E, Ajbro KD, Guitart M, et al: 先天性心疾患および小頭症の患者における新たな間質性10q22-q23重複の微細マッピング
Eur J Med Genet 51: 81–86 (2008).
Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, et al: Bipolar I障害および統合失調症: アシュケナージ・ユダヤ人カセパーレントトリオの64の候補遺伝子の440-一塩基多型スクリーニング Am J Hum Genet 77: 918–936 (2005).
転倒DL:ニューレグリン:機能、形態、およびシグナル伝達戦略。 Cell Exp 284: 14-30 (2003)
Goss PW、Voullaire L、Gardner RJ:染色体内挿入による重複10q22.1eq25.1:2番目の症例。 Ann Genet 41: 161-163 (1998)
Han JY, Kim KH, Jun HJ, Je GH, Glotzbach CD, Shaffer LG: 母体の挿入転座による10番染色体(q22-q24)の部分的トリソミー(15; 10)。 Am J Med Genet A 131: 190–193 (2004).
Hershberger RE、Parks SB、Kushner JD、Li D、Ludwigsen Sら:家族性または特発性拡張型心筋症患者313人からのMYH7、TNNT2、SCN5A、CSRP3、LBD3、およびTCAPで同定されたコード配列変異。 Clin Transl Sci 1: 21–26 (2008).
Kato M, Chou TF, Yu CZ, DeModena J, Sternberg PW: LINKINは細胞接着に必要な新しい膜貫通蛋白質である。 3:e04449 (2014)を参照。
Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, et al: 17q21.31微小欠失症候群の臨床的および分子的描出。 J Med Genet 45: 710-720 (2008)。
Larsen HJ, Howe JR: 若年性ポリポーシス症候群、in Pagon RA、Adam MP、Ardinger HH、Wallace SE、Amemiya A, et al (編): GeneReviews® [Internet] (University of Washington, Seattle 1993-2017)。 初回掲載: 2003年5月13日、最終更新: 2015年12月3日。
Li J, Yen C, Liaw D, Podsypanina K, Bose S, et al: PTEN、ヒトの脳、乳房、および前立腺がんにおいて突然変異した推定タンパク質チロシンホスファターゼ遺伝子。 科学275:1943-1947(1997)。
Lupski JR, Stankiewicz P: ゲノム障害: 再編成と伝達された表現型の分子機構。 PLoS Genet 1:e49 (2005).
Menko FH, Kneepkens CM, de Leeuw N, Peeters EA, Van Maldergem L, et al: PTENおよびBMPR1A遺伝子を含む10q23の微小欠失に関連する可変表現型。 Clin Genet 74: 145-154 (2008)。
Nevado J, Mergener R, Palomares-Bralo M, Souza KR, Vallespin E, et al: New microdeletion and microduplication syndromes: 包括的レビュー。 Genet Mol Biol 37 Suppl 1: 210– 219 (2014).
Petrova E, Neuner C, Haaf T, Schmid M, Wirbelauer J, et al: LCR3/4-隣接10q22.3q23.2の微小欠失およびまれな表現型特徴を有する少年。 モールシンドローム5: 19-24 (2014)
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, et al: 多座位ゲノム変異に起因する疾患表現型の解明。 N Engl J Med 376: 21-31 (2017)
Reddy KS, Mardach R, Bass H: 10q22.1q24.32内の染色体微小欠失のOligoarray (105K) CGH分析。 細胞遺伝学研究132:113-120(2011)。
斎藤H、山村KI、鈴木N:神経堤由来細胞における骨形成蛋白質受容体1A型シグナル伝達の低下は顔面異型を引き起こす。 ディスクモデルMech 5: 948-955 (2012)
Singh S, Aftimos S, George A, Love DR: 10q23.1の間質性欠失および3つの10qdel症候群の確認。 シンガポールメッドJ52:e143-146(2011)
スラボチネックAM:染色体マイクロアレイにより検出された新規微小欠失症候群。 Hum Genet 124: 1-17 (2008)。
van Bon BW, Balciuniene J, Fruhman G, Nagamani SC, Broome DL, et al: 再発性10q22q23欠失および重複の表現型。
Eur J Hum Genet 19: 400–408 (2011).
Vasan RS, Glazer NL, Felix JF, Lieb W, Wild PS, et al: 心臓の構造および機能に関連する遺伝的変異体: ゲノムワイド関連データのメタアナリシスおよび複製。 自工会302:168-178(2009)
Vatta M, Stetson SJ, Perez-Verdia A, Entman ML, Noon GP, et al: 末期心筋症患者におけるジストロフィンの分子リモデリングおよび補助装置療法を受けている患者における回復。 Lancet 359: 936-941 (2002)
Yamazaki M、Araki K、Shibata A、Mishina Mら:マウスグルタミン酸受容体チャネルファミリーの新規メンバーをコードするcDNAの分子クローニング。 Biochem Biophys Res Commun 183: 886–892 (1992).
Zhang W, St-Gelais F, Grabner CP, Trinidad JC, Sumioka A, et al: カイニン酸型グルタミン酸受容体を調節する膜貫通副サブユニット。 ニューロン61:385-396(2009)。
Zhou XP, Waite KA, Pilarski R, Hampel H, Fernandez MJ, et al: Germline PTENプロモーター突然変異およびCowden/Bannayan-Riley-Ruvalcaba症候群における欠失は、異常なPTENタンパク質およびホスホイノシトール-3-キナーゼ/Akt経路の調節異常をもたらす。 Am J Hum Genet 73: 404–411 (2003).